 |
Başlıklar |
|
AKDENİZ ANEMİSİ (BETA TALASEMİ) TAŞIYICILIĞI VE HASTALIĞI
Beta talasemi
anne ve babadan çocuklara kalıtsal olarak geçen, önlenebilir bir kan
hastalığıdır. Türkiye’nin de içinde olduğu Akdeniz ülkelerinde önemli bir halk
sağlığı sorunudur. Taşıyıcıların saptanması, genetik danışma ve doğum öncesi
tanı konabilmesiyle engellenebilir bir hastalık olmasına rağmen, dünyada her yıl
en az 365.000 talasemi hastası doğmakta ve tedavi görmektedir. Türkiye’de
yaklaşık 1.300.000 talasemi taşıyıcısı ve 4.500 kadar talasemi hastası vardır.
Beta talasemi
hastalığı ağır, tedavi düzgün sürdürülmezse yaşam süresini belirgin kısaltan ve
yaşam kalitesini çok olumsuz etkileyen bir hastalıktır. Hastalığın tedavisi
zordur ve maliyeti çok yüksektir. Talasemili bir hastanın yıllık tedavi maliyeti
10.000 dolar civarındadır. Bu nedenle, hastalıklı bireylerin doğmasını
engellemek çok önemlidir ve gerekli koruyucu önlemlerin alınması devlet
tarafından da desteklenmektedir.
Beta
talasemi nasıl oluşur?
Kanımızda
kırmızı kan hücreleri içinde yer alan hemoglobin, dokular için gerekli olan
oksijeni taşır. Hemoglobin molekülünün hem ve globülin olmak üzere iki kısmı
vardır. Sağlıklı bir kişide globin proteini iki çift polipeptid zincirinden
oluşur. Polipeptid zincirlerine göre erişkin bir kişinin eritrositlerinde 3 ayrı
tipte hemoglobin bulunur:
1. Hemoglobin
A: Globin parçası 2 alfa, 2 beta polipeptid zincirinden yapılmıştır. Total
hemoglobinin %96-98’ini içerir.
2. Hemoglobin
F: Globin parçası 2 alfa ve 2 gama polipeptid zincirinden yapılmıştır; total
hemoglobinin %1’inden azını kapsar.
3. Hemoglobin
A2: Globin parçası 2 alfa ve 2 delta polipeptid zincirinden yapılmıştır, total
hemoglobinin %2-3’ünden azını içerir.
Hemoglobin
yapımı genlerin kontrolü altındadır ve ailesel, genetik bir defekt sonucu
hemoglobini oluşturan globin zincirlerinden birinin yapımında yetersizlik veya
bozukluk oluşursa talasemi ortaya çıkar. Globin zincirlerinden hangisi
sentezlenemiyorsa veya hangisinin sentezi azalmışsa talasemi onun adıyla anılır.
Örneğin beta globin sentezindeki değişiklik beta talasemi hastalığına, alfa
globin sentezindeki değişiklik alfa talasemiye neden olur. Beta talasemide
hemoglobin yapısındaki bozukluk sonucu kırmızı kan hücreleri hızla yıkılır ve
bunun sonucunda anemi, yani kansızlık ortaya çıkar.
Beta
talasemide taşıyıcılık ve hastalık nedir?
İnsanlarda bir
özelliğe ait genlerden iki adet bulunur, biri anneden, diğeri babadan geçer.
Beta talasemi için anne ve babadan geçen globin geni normalse çocuk normal, biri
değişikliğe uğramışsa çocuk taşıyıcı, ikisi de değişikliğe uğramışsa çocuk hasta
olur.
Beta
talasemi nasıl ailesel geçiş gösterir?
Bir beta
talasemi taşıyıcısı, taşıyıcı olmayan normal bir kişi ile evlenirse doğacak her
bir çocuk için %50 taşıyıcı, %50 normal olma olasılığı vardır. Bu durumda
hastalık ortaya çıkmaz, korkulacak bir durum yoktur; ancak çocuklarda
taşıyıcılık olup olmadığı araştırılır. Taşıyıcı olanların ileride sağlıklı
çocukları olması için gerekli bilgi verilir, taşıyıcı biri ile evlenirse
çocuklarında hastalık olabileceği anlatılır.
Bir toplumda
taşıyıcılık oranı ne kadar yüksekse rastlantısal olarak iki taşıyıcının evlenme
ve hasta çocuk sahibi olma olasılığı o kadar yüksektir. İki taşıyıcının
evlenmesi sonucunda her bir çocuk için %25 oranında hastalıklı doğma, %50
taşıyıcı olma ve %25 normal doğma ihtimali vardır. Özellikle akraba
evliliklerinde hastalıklı çocuk doğma riski yüksektir, bu kişilerin evlilik
öncesi gereken tetkikleri yaptırmaları çok önemlidir.
Türkiye’de
beta talasemi taşıyıcılığı ne kadardır ve bölgelere göre farklılık gösterir mi?
Ülkemizde beta
talasemi taşıyıcılığı sıklığı %2,1 dolayındadır. Bu sayı farklı bölgelerde
artmakta, taşıyıcılık sıklığı %13’e kadar yükselmektedir (Antalya %13, Edirne
%6,4, Urfa %6,4, Aydın %5,1, Antakya %4,6, İzmir %4,8, Muğla %4,5, İstanbul
%4,5). Akdeniz, Ege ve Trakya bölgeleri taşıyıcılığın yüksek olduğu bölgelerdir.
Talasemi
taşıyıcılığı ve hastalığı nasıl saptanır?
Hasta veya
taşıyıcı olduğu bilinen ailelerde tarama sonucu veya kansızlık nedeniyle
getirilen çocuklarda tanı konur. Taşıyıcı kişiler hafif kansızdır, demir
tedavisinden yarar görmezler. Tam kan sayımının iyi değerlendirilmesi ve
hemoglobin elektroforezi yapılmasıyla tanı kolayca konur. Hasta olanlarda ağır
kansızlık vardır; anne, baba ve çocuğun tam kan sayımı, hemoglobin elektroforezi
ve genetik tetkikleri yapılarak kesin tanı konur.
Beta
talaseminin klinik şekilleri nelerdir?
Beta talasemi
klinik olarak 4 şekilde görülür:
1. Talasemi
major (Ağır hasta tipi): Anne ve baba taşıyıcıdır, çocuğa geçen iki globin geni
de defektlidir. Genellikle bebek 6 aylık olduğunda ağır bir kansızlık ortaya
çıkar ve hayatın ilk 4-12 ayında tanı konur. Halsizlik, solukluk, iştahsızlık,
huzursuzluk, karaciğer, dalak büyümesi sonucu karın şişliği, kemiklerde
genişleme ve incelme, burun kökü basıklığı, alın ve diğer yüz kemiklerinde
çıkıntı ile anormal yüz görünümü ortaya çıkar. Laboratuvar tetkiklerinde
ortalama eritrosit hacmi ve ortalama eritrosit hemoglobin miktarları azalmıştır;
ağır bir anemi vardır. Hemoglobin düzeyi 7 g/dl’nin altındadır. Hemoglobin
elektroforezinde hemoglobin A hemen hemen yoktur ve yerini hemoglobin F
almıştır. Hemoglobin A2 normal, düşük veya hafif artmış olabilir. Bu hastalar
hayatları boyunca düzenli tedavi görmek zorundadırlar.
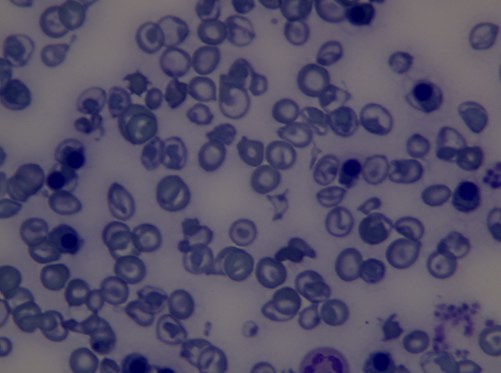
Beta
talasemi majorlu bir hastanın periferik kan yaymasında eritrositlerin görünümü
2. Talasemi
intermedia (Orta ağır hasta): Anne ve baba taşıyıcıdır, çocuğa geçen iki beta
globin geni de defektlidir ancak talasemi majordan farkı genlerdeki değişim daha
hafif bir klinik tabloya yol açan tiptedir. Klinik daha ılımlıdır, kansızlık
daha hafiftir. Hemoglobin düzeyi 7-10 g/dl arasındadır. Hastalar genellikle
düzenli kan transfüzyonu gereksinimi duymazlar.
3. Talasemi
minör (Talasemi taşıyıcılığı): Bu kişilerin hafif derecede kansızlık dışında
sorunları olmaz. Laboratuvar tetkiklerinde hemoglobin değeri hafif düşüktür,
ortalama eritrosit hacmi ve ortalama eritrosit hemoglobin miktarları azalmıştır.
Demir eksikliğinden farklı olarak eritrosit sayıları normal veya artmıştır.
Hemoglobin elektroforezinde hemoglobin A2 ve hemoglobin F hafif yüksektir.
Talasemi taşıyıcılığı bir hastalık değildir ve tedavi gerektirmez. Talasemi
taşıyıcılarında da bebeklik döneminde iyi beslenememe nedeniyle demir eksikliği
gelişebilir. Bu nedenle demir eksikliği tanısı konan hastalar demir tedavisi
sonrasında değerlendirilmeli ve birlikte bulunabilecek talasemi taşıyıcılığı
atlanmamalıdır. Talasemi taşıyıcılarının büyük bir çoğunluğu bu hastalığı
taşıdıklarını bilmezler ancak kendileri gibi taşıyıcı biri ile evlenip hasta bir
çocuk sahibi olduklarında ya da tarama sonrasında öğrenirler.
4. Talasemi
minima: (Talasemi taşıyıcılığı): Bulgular talasemi minördeki gibidir, ancak
hemoglobin elektroforezi normal saptanır, tanı gen analizi ile konur.
Beta
talasemili hastanın tedavi ve izlemi nasıldır?
Beta talasemili
hasta ömür boyu her 3-4 haftada bir kan desteğine ihtiyaç duyar. Talasemili
hastanın hemoglobini 9,5 g/dl’nin üzerinde tutulmalıdır. Kansızlığı düzeltmek
için verilen kan transfüzyonları vücutta demir birikmesine yol açar ve kalp,
karaciğer, tiroid, pankreas ve dalak gibi organlarda hücre hasarına yol açar.
Hastalarda kalp yetmezliği, şeker hastalığı, gelişme geriliği ve hormonal
yetersizlik gibi problemler gelişir. Bunların gelişmemesi için demir birikimini
önlemek amacıyla hastalara genellikle 3 yaş civarında özel bir pompa ile
haftanın en az 5 günü, 8-12 saat süren deri altı infüzyonu ile verilen bir ilaç
(desferrioksamin) başlanır. Son yıllarda ağızdan alınan tablet şeklindeki
ilaçlar da doktorun uygun gördüğü hastalarda kullanılmaya başlanmıştır.
Talasemili
hastalarda tam kan sayımı, kan demir düzeyi, kalp, karaciğer ve hormonal sistem
düzenli olarak değerlendirilir; kan yolu ile bulaşan hastalıklara dikkat edilir.
Yıllık kan tüketimi normalin 1,5 katını aşmışsa ileri yaşlarda dalak operasyonla
çıkartılır. Dalağın çıkarılması kan ihtiyacını azaltır ancak kesin çözüm
değildir.
Kemik iliği
nakli hastalığı tamamen düzeltebilen bir tedavi yöntemidir. Özellikle iyi tedavi
edilen, karaciğerde hasar oluşmamış hastalarda, doku tipi uygun sağlıklı
kardeşten yapılan kemik iliği nakli başarılı olmaktadır. Ancak bazı olgularda
kemik iliği nakli sırasında veya sonrasında çeşitli ciddi problemler ortaya
çıkabilmekte veya nakil başarısızlıkla sonuçlanmaktadır.
Araştırmalara
devam edilen gen nakli henüz hastalara uygulanmamaktadır.
Beta
talasemili hastalarda beslenme nasıl olmalıdır?
Bu bireylerde
verilen kanlarla demir birikmesi yanısıra barsaktan emilen demir miktarı da
arttığından demirden zengin gıdalarla beslenmekten kaçınılmalıdır. Ancak bu
durum talasemi taşıyıcıları için geçerli değildir; talasemi taşıyıcılarında
gereksinimin arttığı durumlarda demir eksikliği anemisi de gelişebilmektedir.
Beta
talasemili hastalar evlenip çocuk sahibi olabilir mi?
Düzenli kan
transfüzyonu ve demir birikimi için tedavi uygulanan hastalar evlenip çocuk
sahibi olabilirler. Beta talasemi hastası olan bir kişi taşıyıcı olmayan,
normal bir kişi ile evlenirse çocukları taşıyıcı olur, hasta olmaz; taşıyıcı bir
kişi ile evlenirse her bir çocuk için %50 hasta, %50 taşıyıcı olma ihtimali
vardır.
Beta
talasemi hastalığından nasıl korunulur?
- Toplum
eğitimi: Toplum beta talasemi konusunda eğitilmeli ve akraba evliliklerinin
riskleri açısından bilgilendirilmelidir.
-
Taşıyıcıların tespiti: Özellikle taşıyıcılığın yüksek oranda görüldüğü
bölgelerde hasta ve taşıyıcı bireylerin tüm akrabalarının kan tetkiki ile
taranması ve evlenecek çiftlerin taşıyıcılık açısından değerlendirilmesi çok
önemlidir.
- Genetik
danışma: Eşlerin ikisinin de taşıyıcı olması durumunda eşlere danışmanlık
verilmeli, genetik tanı merkezlerine yönlendirilmeli ve gebelik öncesinde
gerekli tetkikler tamamlanmalıdır (örnek mutasyon analizi).
-
Prenatal (doğum öncesi) tanı: İki taşıyıcının evliliği söz konusu ise
çiftler mutlaka her gebeliğin erken döneminde (ilk 2 ay) doktora başvurmalı
ve gerekli tetkikleri yaptırmalıdırlar.
Günümüzde prenatal ve preimplantasyon (in vitro fertilizasyon) tanı
yöntemleri ile talasemik hasta çocuğun doğması önlenebilir.
Kaynaklar
- Orkin
SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux SE (Eds). Nathan and
Oski’s Hematology of Infancy and Childhood, Seventh Edition, W.B. Saunders
Company, 2009, Philadelphia, USA.
-
Kliegman RM, Behrman RE, Jenson HB (Eds). Nelson Textbook of Pediatrics, 18th
edition, W.B. Saunders Company, 2007, Philadelphia, USA.
- Canatan
D, Aydınok Y (Eds). Talasemi ve hemoglobinopatiler tanı ve tedavi kitabı,
Talasemi Federasyonu, 2007, Antalya.
-
Yeşilipek A (Ed). Hemoglobinopatiler özel sayısı. Türkiye Klinikleri, 2007,
Ankara.
|


